Do you have a question? Contact us!
"Why are you using CLUSTAL to align?"
Short answer: it works like a charm.
"Clustal is slow!" As you know, in the standard pipeline of the service we do an alignment, a number of models over that alignment, some loop refining and finally a molecular simulation. The tipical processor time consumptions of each step are:
- Clustal: 0.074 seconds per alignment.
- Model: about 30 seconds.
- Loop refining: about 2 minutes per loop, depending on loop sizes.
- Molecular dynamics: about 40 hours using 8 cores, that is 320 hours.
As you may suspect, speed alignment is not our first concern regarding performance. In fact, aligning is the only step of the above that is "synchronous", i.e. you have to wait for it before reach the next page. But, what if alignment speed was a major concern? "I heard somewhere that clustal is orders of magnitude slower than anything else, including hand alignment. Switching to any other algorithm is easy and will payoff". Lets take our 7 hand curated alignment for the inactive GPCR already crystalized, put one of the seven sequences out, 3odu, and realign with:
- Clustal:
clustalo --profile1=6-inactive.aln --profile2=3odu.aln --outfile=clustal_profile.alntook 0.074 seconds. - Mafft:
mafft-linsi --seed 6-inactive.fas 3odu.fas > mafft_profile.fastook 0.047 seconds. - Muscle:
muscle -in 7-inactive.fas -out 7-inactive_2.fastook 0.193 seconds. - Probcons:
probcons 7-inactive.fastook 1.002 seconds. - T_coffee:
t_coffee -profile1 6-inactive.aln -profile2 3odu.aln -out t_coffe_profile.fastook 0.140 seconds.
For 10 repetitions for alignment and averaging times. Seems like clustal is not that bad after all.
"Clustal alignments are bad!" I'm aware that clustal can make big mistakes and produce unusable alignments. But in this particular case it just seems to do a fine work. Lets see the previous generated alignments in the seven alpha helixes. Again, we have the hand curated alignment made from the crystallized structures, without using any alignment program. Note that Muscle or Probcons doesn't accept sequence-to-profile alignments, but all the others do:
- First helix:
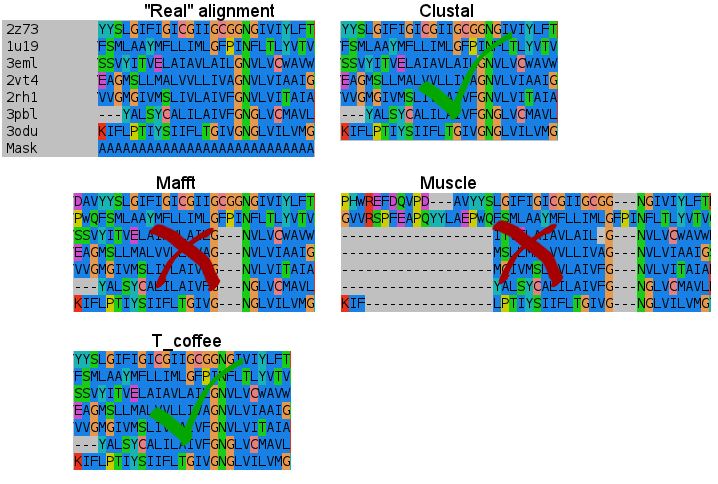
- Second:

- Third:

- Fourth:
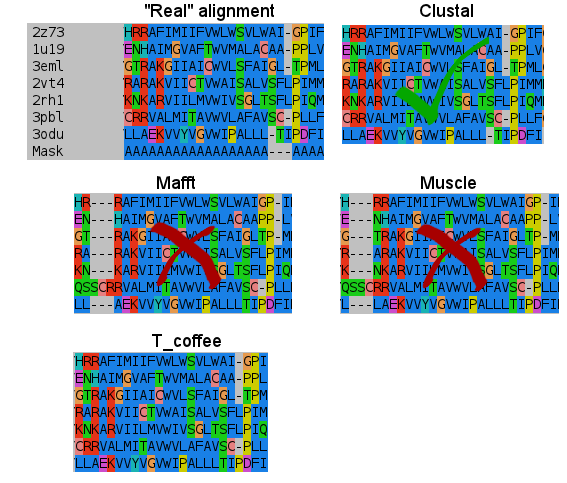
- Fifth:

- Sixth:

- Seventh:

I don't know how Clustal did it, but simply nailed it in the transmembrane regions. T_coffee does a nice work, and also Mafft results are very good. But would you change after seeing this results? (Not shown the Probcons results, as it was tested later. They're in line with Mafft quality, tending to open gaps to fit some residues).
"Your benchmark is bad and you should feel bad!" You're all right to keep doubting about clustal capabilities. That's the reason we offer you the flexibility to edit the clustal alignment. In fact, you're encouraged to do so: no alignments should pass to Modeling stage without deep review by the user. You can also download our profile alignments, align the sequences in your computer with your algorithm of choice and re-upload the alignments back.